Clinical Guidelines for Wolfram Syndrome: A Delphi Consensus Study
Consensus Recommendations for the Clinical Management of Wolfram Syndrome Using a Delphi Method
- Josephine Elliott,*,
- Saumel Ahmadi,*,
- Patrick Yu Wai Man,,,,
- Sarah Gladstone,
- Stephanie Gebel,
- Tracy Lynch,
- Timothy Barrett,#,
- Fumihiko Urano,,#,
- International Wolfram Syndrome Clinical Guidelines Consortium
*Contributed Equally | #Corresponding Authors
Correspondence:
Fumihiko Urano, MD, PhD: 660 South Euclid Ave, St. Louis, Missouri 63110, USA
Email: urano@wustl.edu
Timothy Barrett, MB BS, PhD: Department of Cancer and Genomic Sciences, College of Medicine and Health, Room WXG42, Ground Floor IBR West, The Medical School, Vincent Drive, Birmingham B15 2TT, UK
Email: t.g.barrett@bham.ac.uk
International Wolfram Syndrome Clinical Guidelines Consortium – Members
The following medical and allied health professionals with experience in the clinical care of individuals with Wolfram syndrome contributed to the modified three-round Delphi process by providing feedback, suggestions, and evaluations that helped refine the statements and inform these consensus recommendations.
Consortium Members
- Fumihiko Urano, Washington University, St. Louis, USA
- Timothy Barrett, Birmingham Children’s Hospital and University of Birmingham, UK
- Saumel Ahmadi, Washington University, St. Louis, USA
- Patrick Yu-Wai-Man, University of Cambridge, UK
- Sarah Gladstone, The Snow Foundation, St Louis, USA
- Josephine Elliott, University of Birmingham, UK
- Bess Marshall, Washington University, St Louis, USA
- Alanna Strong, Children’s Hospital of Philadelphia, USA
- Wendy K. Chung, Boston Children’s Hospital and Harvard Medical School, Boston, USA
- Angela M. Reiersen, Washington University, St Louis, USA
- Tamara Hershey, Washington University, St Louis, USA
- Paul F. Austin, Texas Children’s Hospital and Baylor College of Medicine, USA
- Douglas Coplen, St Louis Children’s Hospital and Washington University, St Louis, USA
- Christina Gurnett, Children’s Hospital of Philadelphia and University of Pennsylvania, USA
- Kyle O. Rove, University of Colorado and Children’s Hospital Colorado, USA
- Natasha Leibel, Columbia University, New York, USA
- Yunshuo Tang, Washington University, St Louis, USA
- Louis Philipson, University of Chicago, Chicago, USA
- Rudolph L. Leibel, Columbia University, New York, USA
- Irl B. Hirsch, University of Washington, Seattle, USA
- Gregory Van Stavern, Washington University, St Louis, USA
- Christophe Orssaud, Hôpital Européen Georges Pompidou, Paris, France
- Renuka P. Dias, Birmingham Children’s Hospital and University of Birmingham, UK
- Raniero Chimienti, Vita-Salute San Raffaele University, Milan, Italy
- Agathe Roubertie, Centre Hospitalier Universitaire de Montpellier, Montpellier, France
- Ben Wright, Queen Elizabeth Hospital, Birmingham, UK
- Arunkumar R. Pande, Lucknow Endocrine Diabetes and Thyroid Clinic and Health City Vistaar, Lucknow, India
- Gema Esteban-Bueno, Clinical Management Unit Almería Periférica, Andalusian Health Service, Almería, Spain; Spanish Association for Research and Help to Wolfram Syndrome, Almería, Spain
- Kalpana Suhas Jog, King Edward Memorial Hospital and Research Centre, Pune, India
- Susan Gleeson, Birmingham Children’s Hospital, UK
- Mariam Oniani, M. Iashvili Children’s Central Hospital, Georgia
- Paulina Cruz Bravo, Washington University, St Louis, USA
- Amy Robichaux-Viehoever, Washington University, St Louis, USA
- James Hoekel, Washington University, St Louis, USA
- Leanne Stunkel, Washington University, St Louis, USA
- Lauren Golden, New York University, New York, USA
- Carolina Fischinger Moura de Souza, Casa dos Raros, Porto Alegre, Brazil
- Anne Rochtus, University Hospital Leuven, Belgium
- Esko Wiltshire, University of Otago Wellington and Health New Zealand Capital, Coast and Hutt Valley, New Zealand
- Giulio Frontino, Department of Pediatrics, IRCCS Ospedale San Raffaele, Milano, Italy
- Amelia Caretto, Diabetology and Nutrition Unit, ASST Santi Paolo e Carlo, Milan, Italy
- Denise Williams, NHS England, UK
- Archana Kulkarni, Birmingham Women’s and Children’s Hospital, UK
- Agnieszka Zmysłowska, Medical University of Lodz, Poland
- Sam Peter Gurney, Birmingham Children’s Hospital, UK
- Rinki Murphy, University of Auckland and Auckland Diabetes Centre, Auckland, New Zealand
- Sofia Salahuddin, Queen Elizabeth University Hospital, Birmingham, UK
- Alfredo A. Sadun, Doheny Eye Institute, University of California, Los Angeles, USA
- Marie McGee, Birmingham Women’s and Children’s NHS Trust, UK
- Kristin A. Maloney, University of Maryland School of Medicine, Baltimore, USA
- Stephen I. Stone, Washington University, St Louis, USA
- Yukio Tanizawa, Yamaguchi University, Japan
- Stacy Hurst, Washington University, St. Louis, USA
- Ashley Raterman, Washington University, St. Louis, USA
- Marcela Votruba, Cardiff University and University Hospital Wales, UK
- Leonardo Caporali, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- Claudio Fiorini, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- Raoul Kanav Khanna, Bretonneau University Hospital and INSERM iBraiN U1253, Tours, France; University of Cambridge and Cambridge University Hospitals, Cambridge, UK
- Chiara La Morgia, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy; University of Bologna, Italy
- Susan Patricia Mollan, Queen’s University, Kingston, Ontario, Canada
- An-Guor Wang, Taipei Veterans General Hospital, Taiwan
- Piero Barboni, San Raffaele University, Milan, Italy
- Alina Radu, Hôpital Européen Georges Pompidou and Université Paris Cité/Assistance Publique-Hôpitaux de Paris, Paris, France
- Jacques Beltrand, Hôpital Necker-Enfants Malades and Université Paris Cité/Assistance Publique-Hôpitaux de Paris, Paris, France
- Neringa Jurkute, Moorfields Eye Hospital NHS Foundation Trust, London, UK; UCL Institute of Ophthalmology, London, UK; The National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK
- Emilio Hideyuki Moriguchi, Universidade Federal do Rio Grande do Sul, Brazil
- Elmarie van der Merwe, Birmingham Children’s Hospital, UK
- Julia Rohayem, Ostschweizer Kinderspital St. Gallen, Switzerland
- Mars Skae, Royal Manchester Children’s Hospital, UK
- Andrew R. Lee, Washington University, St Louis, USA
- Andrew Hattersley, University of Exeter, UK
- Maggie Shepherd, Royal Devon University Healthcare NHS Foundation Trust and University of Exeter, UK
- Kashyap Patel, University of Exeter, UK
- Kevin Colclough, University of Exeter, UK
- Valerio Carelli, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy; University of Bologna, Italy
- Michele Carbonelli, University of Bologna, Italy
- Orit Pinhas-Hamiel, Pediatric Endocrine and Diabetes, Sheba Medical Center, Ramat Gan, Israel; Gray Faculty of Medical and Health Sciences, Tel Aviv University, Tel Aviv, Israel
- Noga Minsky, Division of Endocrinology, Diabetes and Metabolism, Sheba Medical Center, Ramat Gan, Israel; Gray Faculty of Medical and Health Sciences, Tel Aviv University, Tel Aviv, Israel
- Odelia Chorin, The Danek Gertner Institute of Human Genetics, Sheba Medical Center, Ramat Gan, Israel; Gray Faculty of Medical and Health Sciences, Tel Aviv University, Tel Aviv, Israel
- Gil Leibowitz, Endocrinology and Metabolism Service, Hadassah Medical Center, Jerusalem, Israel; Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem, Israel
- Avivit Cahn, Internal Medicine Department A, Hadassah Hebrew University Hospital, Jerusalem, Israel; The Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem, Israel
- Stephanie Gebel, The Snow Foundation, St. Louis, USA
- Tracy Lynch, Wolfram Syndrome UK, UK
- Nolwen Le Floch, Association du Syndrome de Wolfram, France
Abstract
Background
Wolfram syndrome is a rare neurodegenerative disorder, most commonly caused by pathogenic variants in WFS1, while cases due to CISD2 are exceedingly rare. The estimated prevalence is 1 in 160,000 to 770,000 individuals worldwide. In these clinical guidelines, disorders caused by WFS1 are referred to as WFS1-Wolfram syndrome, and those caused by CISD2 as CISD2-Wolfram syndrome. Historically, it has been characterized by early-onset, antibody-negative, insulin-dependent diabetes mellitus, progressive optic atrophy, sensorineural hearing loss, arginine vasopressin deficiency, and brainstem and cerebellar atrophy. More recently, partial and late onset forms have been identified. There are currently no licensed disease-modifying treatments, and international clinical guidelines have not previously been established.
Methods
An international steering committee systematically reviewed 273 peer-reviewed publications and generated draft consensus statements across six clinical domains. These statements were evaluated by international specialists in endocrinology, clinical genetics, neurology, ophthalmology and neuro-ophthalmology, psychiatry, and urology, drawn from North America, Europe, Latin America, Oceania, and Asia, using a modified three-round Delphi process. Additional feedback was incorporated from nurses specializing in multidisciplinary Wolfram syndrome care, from leaders of international patient organizations, and from specialists in the genetic diagnosis of monogenic diabetes. Structured feedback from patients and families was gathered through multiple international patient advocacy organizations. Consensus was defined as ≥80% agreement.
Results
All 35 final consensus statements reached the pre-specified consensus threshold of ≥80% agreement, spanning diagnosis and genetic testing, multidisciplinary care organization, neuro-ophthalmology, neurology, endocrinology, urology, gastroenterology, and psychiatry.
Conclusions
These guidelines are the first international clinical consensus for Wolfram syndrome and provide actionable recommendations for clinicians worldwide. Implementation should be accompanied by a prospective audit to expand the evidence base and support future iterations.
Introduction
Wolfram syndrome (OMIM #222300; also known as DIDMOAD: Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, Deafness) is a rare monogenic, multisystem neurodegenerative disease first described by Wolfram and Wagener in 1938 in four siblings presenting with juvenile-onset diabetes mellitus and progressive optic nerve atrophy1. The disease is caused predominantly by biallelic loss-of-function variants in the WFS1 gene, which encodes Wolframin, a transmembrane glycoprotein of the endoplasmic reticulum (ER) critical for ER calcium homeostasis, the unfolded protein response, and pancreatic β-cell survival2,3. A small number of patients have pathogenic variants at a second locus, CISD2, which is associated with additional gastrointestinal symptoms and platelet disorders4.
Over the past three decades, the clinical phenotype of Wolfram syndrome has expanded considerably. In addition to diabetes mellitus and optic atrophy, individuals develop sensorineural deafness, arginine vasopressin deficiency (diabetes insipidus), progressive brainstem and cerebellar neurodegeneration, cerebellar ataxia, autonomic failure, psychiatric illness (depression and psychosis), and neurogenic bladder5. There are no approved disease-modifying treatments. Premature death has been reported at a median age of approximately 30 years, most commonly from brainstem failure and respiratory compromise5. However, there are affected people living into their seventh decade6. In addition, dominantly inherited, partial phenotypes have also been described7-9.
Despite its clinical complexity and global distribution, no international clinical guidelines for Wolfram syndrome have previously existed. Individual clinicians and centers have developed local protocols in isolation, resulting in marked heterogeneity in diagnostic approaches, monitoring schedules, and management practices. Prior to this work, the only consensus documents published for Wolfram syndrome were limited in scope, lacked a formal multi-round methodology, and did not incorporate patient and family perspectives.
Here, we present the first international consensus statement for the diagnosis and clinical management of Wolfram syndrome, developed through a modified three-round Delphi process. The structure and precedent for this work follows the approach established for Cornelia de Lange syndrome, another rare autosomal recessive multisystem genetic disease10, Duchenne muscular dystrophy11, pituitary adenoma12, Beckwith–Wiedemann syndrome13, spinal muscular atrophy14, and Alström syndrome15. These guidelines address the full clinical spectrum of Wolfram syndrome – molecular diagnosis, multidisciplinary care coordination, and organ-specific management – providing actionable recommendations for clinicians worldwide.
Methods
Steering committee and systematic review
An international steering committee was established in December 2024, co-chaired by Fumihiko Urano (Washington University, St. Louis, USA) and Timothy Barrett (University of Birmingham, Birmingham, UK). The committee comprised eight members (four in the United States, four in the United Kingdom), including geneticists, an endocrinologist, a neurologist, a neuro-ophthalmologist, pediatricians, and members of two patient advocacy organizations (one from the United States and one from the United Kingdom). All physicians on the steering committee had clinical experience actively managing at least 20 patients with Wolfram syndrome. The committee conducted a structured PubMed review of 273 peer-reviewed English-language articles published from 1996 through early 2026, covering Wolfram syndrome type 1 and type 2, WFS1-related disorders, Wolfram-like syndrome, DIDMOAD, and the WFS1 Spectrum disorder in adults and children. Pre-clinical studies, review articles, editorials, conference abstracts, and non-English articles were excluded. References were collated in Covidence, and three steering committee members reviewed each article for inclusion based on clinical relevance to diagnosis and management. The committee then met every two weeks over six months to draft 41 candidate consensus statements across six clinical domains: diagnosis and general management; neuro-ophthalmology; neurology; endocrinology; urology and gastroenterology; and psychiatry and cognition.
Expert panel
An international panel of clinicians was recruited to participate in the Delphi process. Panelists were selected on the basis of recognized expertise in Wolfram syndrome or the relevant clinical subspecialty. Specialists were drawn from North America, Europe (United Kingdom, France, Germany, Italy, Denmark, Spain, Belgium, Poland, Switzerland), Georgia, Latin America (Brazil), Oceania (New Zealand), and Asia (Japan, India, Taiwan). The panel spanned the relevant clinical disciplines, including endocrinology, clinical genetics, neurology, ophthalmology and neuro-ophthalmology, psychiatry, urology, pediatrics, and family medicine, together with allied health professionals comprising clinical nurse specialists, clinical psychologists, and a transition coordinator. Panelists’ identities were masked to reviewers during voting rounds.
Delphi process
A modified three-round Delphi methodology was adopted because it is well-suited to rare diseases such as Wolfram syndrome16: it systematically integrates anonymous feedback from international experts and refines candidate statements through iterative steering committee meetings and topic-focused expert sub-group discussions. In each round, panelists rated their level of agreement with each statement on a five-point Likert scale (1 = strongly disagree; 5 = strongly agree) and were able to provide free-text commentary. Panelists were instructed not to rate statements outside their area of clinical expertise. Consensus was defined a priori as agreement (Likert score of 4 or 5) by ≥80% of respondents, in keeping with the upper end of the accepted threshold for Delphi methodology in healthcare. Statements that failed to reach this threshold were revised, merged, or removed based on respondent comments and recirculated in the subsequent round. Up to three rounds of voting were performed; Rounds 1, 2, and 3 received responses from 56, 37, and 32 panelists, respectively. All 35 final consensus statements reached the pre-specified threshold (Figure 1).
Patient consultation
To ensure that the final guidelines were patient-centered, a parallel patient and family survey was conducted through international advocacy organizations: the Snow Foundation, the Ellie White Foundation, the Unravel Wolfram Syndrome Fund, Be A Tiger Foundation, the Eye Hope Foundation, the Ontario Wolfram League, the Gentian Association, the Alliance of Families Affected by Wolfram Syndrome, the Wolfram Heroes Association, Wolfram Syndrome UK, and the Association du Syndrome de Wolfram. Patients and families are uniquely positioned to identify priorities, lived-experience challenges, and outcomes that matter most in everyday life. Twenty-four lay-language items, thematically aligned with the specialist statements, were rated on the same five-point Likert scale, with free-text commentary invited for each statement. The survey was distributed after Round 2 of the specialist Delphi, allowing candidate statements to first be refined by clinical expert input and then re-examined through the lens of patient and family priorities. Responses were analyzed by the steering committee and used to refine the wording, scope, and emphasis of statements before the final specialist round (Round 3). Patient feedback informed the diagnostic classification, monitoring frequency, and patient-centered management priorities for the most disabling symptoms.
Evidence grading system
- B
- Evidence from well-designed observational studies (cohort or case-control studies), or from systematic reviews of such studies. The body of evidence is sufficient to support the recommendation but is not derived from randomized controlled trials.
- C
- Evidence from case series, individual case reports, or expert opinion supported by mechanistic or physiological rationale. The evidence base is limited in quantity or quality.
- Expert opinion
- Recommendation based on the formal consensus of the international expert panel in the absence of sufficient published evidence to assign a higher grade. These recommendations reflect current best clinical practice as agreed by specialists with direct experience managing Wolfram syndrome.
- Patient feedback
- Recommendation shaped by structured input from patients and families through international advocacy organizations (see Patient consultation section for the full list of participating organizations). Patient perspectives were formally integrated into the Delphi process through parallel rounds of voting.
- Delphi Round
- The round in which the statement first reached the pre-specified consensus threshold of ≥80% agreement. Statements reaching consensus in Round 1 were accepted as final without further iteration; those reaching consensus in Rounds 2 or 3 were revised in response to panelist feedback before re-voting.
No statement could be assigned Level A, as there are no randomized controlled trials or meta-analyses of randomized controlled trials in Wolfram syndrome. The absence of randomized controlled trial data to inform clinical practice is itself a key finding of this work and represents an important unmet need.
Results
The consensus panel comprised specialists from multiple clinical and allied health disciplines who participated in the Delphi process conducted from July 2025 through April 2026. Participants completed three rounds of voting and were drawn from the clinical specialties and allied health professions described in the Methods.
In Round 1, 41 statements reached consensus. Following review of the panelists’ free-text comments, 26 statements were revised and three were removed, resulting in 12 finalized statements. The 26 revised statements were carried forward to Round 2, where all achieved consensus. Based on additional written feedback, 13 statements were further revised and two were removed, increasing the total number of finalized statements to 23. Following additional feedback from patient surveys, three of these 13 revised statements were modified before Round 3. All 13 revised statements reached consensus in the final round; however, one statement was removed based on panelists’ written comments, resulting in 34 finalized statements. After completion of Round 3, one additional statement was revised in response to panel feedback, yielding a final set of 35 consensus statements (Figure 1).
Similarly designed statements also reached consensus among patient and family respondents. Patient feedback primarily helped shape the definition of Wolfram syndrome, frequency of clinical assessments, and patient-centered management priorities for the most disabling symptoms.
Recommendations by Clinical Domains
Diagnosis and genetic testing
Statement 1
Wolfram syndrome is caused by biallelic pathogenic variants in WFS1 or CISD2. Diagnosis requires consideration of both clinical pattern and genetic test results, and must be discussed with a medical geneticist and/or genetic counselor. Four clinical subtypes are defined: (A) WFS1-Wolfram syndrome childhood onset; (B) WFS1-Wolfram syndrome adult onset; (C) WFS1-related disorder; (D) CISD2-Wolfram syndrome (consensus ≥80%; level of evidence: C / expert opinion; Round 2).
The WFS1 gene on chromosome 4p16 encodes Wolframin, a 100-kDa transmembrane protein with nine transmembrane domains that localizes to the endoplasmic reticulum (ER) and is expressed at highest levels in the brain, pancreatic β cells, and heart3,17-20 6,21-24. Biallelic loss-of-function variants cause chronic ER stress, activation of the unfolded protein response, mitochondrial dysfunction, and progressive neuronal and β-cell death25. Genotype–phenotype correlations exist: biallelic truncating variants are generally associated with more severe, earlier-onset disease, whereas missense combinations may produce milder phenotypes6. The vast majority of patients with Wolfram syndrome carry pathogenic variants in the WFS1 gene; in these guidelines we refer to this disease as WFS1-Wolfram syndrome. The CISD2 gene on chromosome 4q24 encodes ERIS (NAF-1/miner1), a zinc-finger ER outer-membrane protein involved in iron–sulfur cluster transfer and mitochondrial function4,26-28. Biallelic CISD2 variants cause CISD2-Wolfram syndrome with additional gastrointestinal ulceration and platelet defects4,29. A spectrum of dominantly inherited WFS1-related disorders also exists, ranging from isolated low-frequency sensorineural hearing loss7, isolated optic neuropathy30, isolated cataracts31, combinations of hearing loss and optic neuropathy8,32,33 to combinations of diabetes mellitus, optic atrophy, cataracts, hypotonia and developmental delay9. These autosomal-dominant WFS1-related disorders are related to, but clinically distinct from, autosomal-recessive Wolfram syndrome and are beyond the scope of the present guidelines.
The original description by Dr. Wolfram in 1938 was of four siblings with an inherited association between juvenile-onset diabetes mellitus and progressive optic nerve atrophy1. This combination has since become embedded in the medical literature as clinically diagnostic of Wolfram syndrome5,34.
Since this original description, it has become apparent that there is a spectrum of conditions associated with pathogenic variants in two genes, WFS1 and CISD2. Within the condition we identify as Wolfram syndrome, there is a wide spectrum of severity. We therefore recommend that clinicians consider both the clinical pattern and the genetic test results in order to make a diagnosis; and that a diagnosis should only be made in discussion with a medical geneticist and/or genetic counselor.
We have categorized conditions caused by variants in WFS1 and CISD2 genes into the following sub-categories under the umbrella of Wolfram Spectrum Disorders:
- A-WFS1-Wolfram syndrome childhood onset
- B-WFS1-Wolfram syndrome adult onset
- C-WFS1-related disorder
- D-CISD2-Wolfram syndrome
Some WFS1 variants have been associated with specific patterns such as neonatal diabetes, congenital hearing loss, and hypotonia9, isolated non-syndromic deafness7,35, isolated optic neuropathy30, and isolated cataracts31. These are associated with specific genotypes and are beyond the scope of this guideline.
- To make a diagnosis of Wolfram syndrome childhood onset, the patient must have childhood onset (under 18 years) diabetes mellitus and optic atrophy, together with biallelic pathogenic/likely pathogenic variants in the WFS1 gene (autosomal recessive).The presence of one pathogenic/likely pathogenic variant in the WFS1 gene and one variant of unknown significance may confirm a diagnosis of Wolfram syndrome if the clinical picture is consistent with Wolfram syndrome, and these findings must be discussed with a medical geneticist and/or genetic counselor.
- To make a diagnosis of Wolfram syndrome adult onset, the patient will likely have adult onset (18 years or older) diabetes mellitus and optic atrophy, together with biallelic pathogenic/likely pathogenic variants in the WFS1 gene (autosomal recessive).The adult-onset form has been separated from the childhood onset form as it usually has a much slower time-course, and can have milder features. The adult form is usually caused by missense variants in the WFS1 gene, for instance the variant commonly seen in the Ashkenazi Jewish population36-38.
- To make a diagnosis of WFS1-related syndromic disorder, the affected person must have profound early-onset deafness with or without optic atrophy, together with one pathogenic / likely pathogenic variant in the WFS1 gene occurring either as an autosomal dominant or as a de novo variant.
- To make a diagnosis of CISD2 Wolfram syndrome, the patient must have childhood onset (under 18 years) diabetes mellitus and optic atrophy, gastrointestinal symptoms of ulceration and a platelet disorder, together with biallelic pathogenic/likely pathogenic variants in the CISD2 gene (autosomal recessive)4.
Disclaimer: Autosomal dominant WFS1-related disorders exist along a broad spectrum of presentation. There is a category of WFS1-related non-syndromic disorders in which individuals may show isolated findings, such as isolated sensorineural hearing loss, isolated optic neuropathy, or isolated cataracts. Although there is one known dominant WFS1 variant associated with isolated diabetes, in most cases WFS1 dominant variants do not cause monogenic diabetes39,40. Caution is needed, as some individuals who initially appear to have “non-syndromic” forms may later develop additional features. In addition, patients who seem to have WFS1-related syndromic disorders may develop certain symptoms for reasons unrelated to their WFS1 variants, such as autoimmune type 1 diabetes or antibody-negative type 1 diabetes41. Therefore, for each symptom commonly seen in Wolfram syndrome and WFS1-related disorders, other potential causes should be carefully ruled out before concluding that the symptom is WFS1-related. The severity, symptoms, and progression vary among individuals. Genetic results should always be interpreted with a qualified health professional, such as a medical geneticist or genetic counselor, as interpretation without appropriate expertise may be misleading.
Multidisciplinary care organization
Statement 2
Patients aged under 18 years should be offered an annual multidisciplinary assessment, or sooner if there is significant deterioration. Patients aged 18 years or older should be offered assessment every 1–2 years depending on disease severity, or sooner if there is significant deterioration (consensus ≥80%; expert opinion / patient feedback; Round 3).
The multisystem nature of Wolfram syndrome necessitates coordinated care from multiple specialties2,5. The panel recommends that all patients be assessed by a formal multidisciplinary team (MDT) at regular intervals: annually for patients under 18 years of age and every 1–2 years for adults (depending on disease severity and symptom burden), with earlier review following significant clinical deterioration. The judgment as to what constitutes significant clinical deterioration, and the decision to bring forward a multidisciplinary review, is best made by the patient’s primary or local specialist team, who can coordinate timely assessment without displacing the routine review of other patients.
Statement 3
A core Wolfram syndrome multidisciplinary team will ideally include a coordinator (geneticist or nurse navigator), endocrinologist, neurologist, ENT/audiology specialist, ophthalmologist or neuro-ophthalmologist with optometry support, and urologist with access to urodynamics. Additional members as clinically indicated may include a genetic counselor, clinical psychologist or psychiatrist, speech and language therapist, physical therapist, gastroenterologist, adult-care transition coordinator, patient support group family liaison, and sleep specialist (consensus ≥80%; expert opinion / patient feedback; Round 3).
A core Wolfram syndrome MDT should include a coordinator (medical geneticist or nurse navigator), endocrinologist, neurologist, ENT/audiology specialist, ophthalmologist with optometry support, and urologist with access to urodynamic testing. Additional members, including a clinical psychologist/psychiatrist, a genetic counselor, a speech and language therapist, a physical therapist, a gastroenterologist, an adult-care transition coordinator, a patient advocacy liaison, and a sleep specialist, should be included according to individual clinical need and local availability. Given the substantial psychological and neuropsychiatric burden of Wolfram syndrome, access to clinical psychology or psychiatry is particularly important and should be considered a key component of the team wherever possible.
Statement 4
Upon diagnosis, all patients should be offered contact with a patient advocacy organization and assessment for entitlement to benefits, caregiver support, and (for children or young people) educational support (consensus ≥80%; expert opinion; Round 1).
Patient advocacy organizations are integral partners in care and a valuable source of practical, emotional, and educational support from the time of diagnosis. All patients and families should be signposted to relevant organizations (Table 1) and assessed for entitlement to benefits and caregiver support, with educational support arranged for children and young people.
Statement 5
Transition from pediatric to adult care should begin early in the second decade of life. The young person and care providers should both be involved in planned transfer to adult services (consensus ≥80%; expert opinion; Round 1).
At diagnosis, all patients and families should be offered contact with patient advocacy organizations and assessment for entitlement to benefits, caregiver support, and educational support. Transition from pediatric to adult care should begin in the second decade of life, with active involvement of the young person in all planning discussions. Use of a structured transition readiness tool, such as the Transition Readiness Assessment Questionnaire42 or the Ready Steady Go programme43, can help guide and document this process.
Neuro-ophthalmology
Statement 6
Patients with a Wolfram Spectrum Disorder should have an ophthalmological assessment at least once a year, including best-corrected visual acuity (cycloplegic refraction in children), color vision, intraocular pressures, slit-lamp examination of the anterior and posterior segments, visual fields (if age-appropriate), and OCT imaging of the optic nerve and macula (if age-appropriate) (consensus ≥80%; expert opinion; Round 2).
Progressive optic atrophy typically presents in the first or second decade of life and progresses to legal blindness in most patients. Visual loss begins with deficits in color vision and central visual field defects. Optical coherence tomography demonstrates early retinal nerve fiber layer thinning, which can precede visual symptoms.
Progressive optic atrophy in Wolfram syndrome causes bilateral central visual field loss, leading to legal blindness in the majority of patients44-46. Annual ophthalmological assessment is recommended for all patients and should include best-corrected visual acuity (with cycloplegic refraction in children), color vision, intraocular pressures, slit-lamp examination of the anterior and posterior segments, visual fields (if age-appropriate), and optical coherence tomography (OCT) of the optic nerve and macula (if age-appropriate). Subclinical retinal nerve fiber layer thinning may precede symptomatic visual loss and is best detected with OCT imaging; macular ganglion cell layer measurements on OCT may precede retinal nerve fiber layer changes.
Statement 7
There is no proven treatment that can slow the progression of visual loss in Wolfram Spectrum Disorders. Management remains supportive, including referral to a low-vision clinic where available, registration as severely sight impaired (if applicable in country), occupational rehabilitation, and special educational needs support (consensus ≥80%; expert opinion; Round 2).
There is currently no proven treatment to slow the progression of visual loss in Wolfram Spectrum Disorders. Management therefore remains supportive. Referral to low vision services should be made promptly when functional impairment arises. Depending on national systems, patients should be registered as severely sight impaired, referred for occupational rehabilitation, and assessed for special educational needs as appropriate. Certification of sight loss, where available, can provide access to additional services and concessions that may benefit the patient.
Neurology
Statement 8
At their annual neurology review (or every 2 years if stable), all patients should be assessed for mental status and cognition (2-step commands, serial 7s, reading, repetition; MoCA/Blind-MoCA or neuropsychology evaluation if abnormal); cranial nerve function (smell, taste, dysarthria, facial symmetry, swallowing or choking difficulties, apneic episodes); motor function (tone, power); sensory function (vibration, temperature, pin-prick); reflexes; coordination; and gait. Speech-language therapy, occupational therapy, and physiotherapy should be considered (consensus ≥80%; expert opinion; Round 2).
Progressive neurodegeneration is the hallmark of later-stage disease and the primary determinant of mortality, and assessment should begin with clinical examination before neuroimaging. Magnetic resonance imaging (MRI) demonstrates progressive atrophy of the brainstem, cerebellum, thalamus, and basal ganglia47-49. Clinical manifestations include cerebellar ataxia, dysarthria, dysphagia, autonomic dysfunction, and ultimately respiratory failure5.
Neurological involvement in Wolfram syndrome is progressive, diverse, and frequently underrecognized. Annual neurological review (or every two years if clinically stable) should include systematic assessment of cognition, cranial nerve function, motor function, sensory function, coordination, reflexes, and gait. Speech-language therapy, occupational therapy, and physiotherapy should be considered at each review.
Statement 9
Patients with imbalance should be referred to a physiotherapist or physical therapist to assess gait, fall-risk, and learn balance exercises. If vestibular dysfunction is suspected, refer to ENT, occupational therapy, and/or audiology (consensus ≥80%; expert opinion; Round 2).
Specific neurological complications require targeted management. Patients with balance impairment should be referred to physiotherapy for gait assessment, fall-risk stratification, and exercise prescription. Vestibular dysfunction warrants ENT referral. Autonomic dysfunction, evidenced by orthostatic hypotension and temperature dysregulation, should be assessed annually by measurement of lying and standing blood pressure. Chronic fatigue should be systematically assessed using a validated questionnaire, such as the Fatigue Severity Scale50, at each clinic visit.
Statement 10
Patients diagnosed early in life should be assessed for hearing loss every 0.5–1 year for the first 4–5 years of life: auditory brainstem responses (or evoked otoacoustic emissions if unavailable); assessment for hearing aids; audiogram, speech discrimination tests, and tympanometry (age-dependent). After the first 5 years of life, review hearing at a minimum of every 2 years, with earlier reassessment if acute deterioration, device change, or key school transition (consensus ≥80%; expert opinion / patient feedback; Round 3).
Sensorineural deafness is present in approximately 60–70% of patients, predominantly affecting high frequencies51-53. Assessment and appropriate amplification are important determinants of speech development and quality of life.
Hearing assessment is critical, particularly in young children, as early intervention with hearing amplification is an important determinant of speech and language development; cochlear implantation is generally reserved for patients with profound early-onset (autosomal dominant) deafness rather than for childhood- or adult-onset Wolfram syndrome. For patients diagnosed in infancy, formal audiological testing (rather than caregiver report alone) every 6–12 months for the first 4–5 years of life is recommended, with biennial review thereafter once hearing is stable. Swallowing assessment by a speech and language specialist is indicated if dysphagia, dysarthria, weight loss, recurrent chest infections, or prolonged mealtimes are reported.
Statement 11
If there are concerns for dysphagia, dysarthria, or features suggestive of swallowing difficulties (weight loss, recurrent chest infections, choking, prolonged mealtimes), patients should be assessed by a speech and language specialist, gastroenterologist, or other relevant specialist per local standard of care (consensus ≥80%; expert opinion; Round 2).
Swallowing difficulties may reflect progressive brainstem and cranial nerve involvement and carry a risk of aspiration and malnutrition5,54. Where dysphagia, dysarthria, weight loss, recurrent chest infections, choking, or prolonged mealtimes are reported, prompt assessment by a speech and language specialist, gastroenterologist, or other relevant specialist should be arranged according to local standards of care55.
Statement 12
If symptoms or signs suggestive of sleep apnea are present (morning headaches, snoring, excessive daytime sleepiness), patients should be assessed with polysomnography (consensus ≥80%; expert opinion; Round 1).
Sleep-disordered breathing, including central and obstructive sleep apnea, may arise from brainstem involvement and contribute to fatigue, morning headaches, and daytime sleepiness. Where suggestive symptoms or signs are present, polysomnography should be undertaken and treatment instituted as indicated56.
Statement 13
Patients may experience autonomic dysfunction including temperature dysregulation and fluctuating blood pressure. Orthostatic vitals should be checked at least annually: heart rate and blood pressure should be measured after 5 minutes supine, then after standing for 1 minute and 3 minutes (consensus ≥80%; expert opinion / patient feedback; Round 3).
Autonomic dysfunction is common and may manifest as orthostatic intolerance, temperature dysregulation, and fluctuating blood pressure. Orthostatic vital signs should be checked at least annually, with heart rate and blood pressure measured after 5 minutes supine and again after standing for 1 and 3 minutes, and symptomatic postural hypotension managed with conservative measures in the first instance57.
Statement 14
Chronic fatigue is common and should ideally be assessed with a validated questionnaire (e.g., Fatigue Severity Scale) at clinic appointments. Management includes screening for and treating underlying causes (anxiety, depression, anemia, sleep disorders, thyroid disease), patient information, sleep hygiene, hydration and nutrition, and supervised graded aerobic exercise. Referral to physiotherapy or occupational therapy should be considered where available (consensus ≥80%; expert opinion / patient feedback; Round 3).
Chronic fatigue is a frequent and disabling symptom that significantly affects quality of life. Assessment with a validated tool such as the Fatigue Severity Scale helps quantify severity and monitor change50. Management should include screening for and treating contributory factors such as anxiety, depression, anemia, sleep disorders, and thyroid disease, alongside advice on sleep hygiene, hydration, nutrition, and supervised graded aerobic exercise, with referral to physiotherapy or occupational therapy where available.
Statement 15
Migraines and headaches are common in Wolfram syndrome and can be exacerbated by systemic infections. Reversible causes should be screened for and treated (dehydration, infection, poor glycemic control, poor sleep hygiene). Acute and preventative management depends on headache type, severity, and age (consensus ≥80%; expert opinion; Round 2).
Headaches and migraines are common in Wolfram syndrome and may be exacerbated by systemic infections, dehydration, poor glycemic control, and poor sleep58,59. Reversible triggers should be identified and addressed before escalating to specialist headache management. Brain MRI without contrast, with thin cuts through the brainstem and orbits, should be obtained at diagnosis and when new or worsening neurological impairment is present. Movement disorders including ataxia, tremors, and parkinsonism may be manifestations of Wolfram syndrome-related neurodegeneration but may also reflect co-existing pathology and should prompt neurological review.
Statement 16
MRI brain without contrast, with thin cuts through the brainstem and orbits, should be considered in patients with a new diagnosis of Wolfram syndrome or with new or worsening neurological impairment. Wolfram syndrome patients show progressive atrophy of the cerebellum, brainstem, thalamus, and/or basal ganglia (consensus ≥80%; expert opinion; Round 3).
Brain MRI without contrast, with thin cuts through the brainstem and orbits, supports diagnosis and characterization of neurodegeneration. Imaging typically demonstrates progressive atrophy of the cerebellum, brainstem, thalamus, and basal ganglia47,49. MRI should be considered at new diagnosis and when new or worsening neurological impairment is identified.
Statement 17
Movement disorders (ataxia, tremors, myoclonus, parkinsonism, tics) may be present in Wolfram syndrome, but other underlying diagnoses may co-exist and should be considered as necessary (consensus ≥80%; expert opinion / patient feedback; Round 3).
Movement disorders, including ataxia, tremor, myoclonus, parkinsonism, and tics, may occur as part of Wolfram syndrome-related neurodegeneration. Because co-existing or alternative diagnoses can produce similar features, these presentations should prompt neurological review and consideration of other underlying causes where clinically appropriate5,60.
Statement 18
Seizures are not common in Wolfram syndrome. If they occur, investigations should be undertaken to establish the underlying cause (consensus ≥80%; expert opinion; Round 2).
Seizures are uncommon in Wolfram syndrome. When they do occur, they should not be assumed to be a direct manifestation of the underlying disorder, and appropriate investigation should be undertaken to establish and treat the underlying cause61.
Endocrinology
Statement 19
To diagnose diabetes mellitus in Wolfram syndrome, follow ISPAD clinical criteria: (1) classic symptoms or hyperglycemic crisis with plasma glucose ≥11.1 mmol/L (200 mg/dL); or (2) fasting plasma glucose ≥7.0 mmol/L (125 mg/dL); or (3) 2-hour post-load glucose ≥11.1 mmol/L during OGTT (200 mg/dL); or (4) HbA1c ≥48 mmol/mol (6.5%). In the absence of unequivocal hyperglycemia, two abnormal test results are required. The absence of islet cell antibodies at diagnosis raises suspicion that diabetes mellitus is not type 1 (consensus ≥80%; level B; Round 2).
Diabetes mellitus is the presenting feature in most patients, typically appearing in the first decade of life (median onset 6 years)2,6,34,62. It is characterized by insulin deficiency, the absence of islet autoantibodies, and sometimes residual C-peptide more than a year after diagnosis, features that distinguish it from type 1 autoimmune diabetes and are critical for correct diagnosis.
Diabetes mellitus in Wolfram syndrome is insulin-deficient but distinct from type 1 autoimmune diabetes. Diagnosis follows established International Society for Pediatric and Adolescent Diabetes (ISPAD) criteria63. Critically, the absence of islet autoantibodies at diagnosis should prompt consideration of Wolfram syndrome and genetic testing in any child with unexplained insulin-requiring diabetes.
Statement 20
Where feasible, continuous glucose monitoring as part of a hybrid closed-loop insulin delivery system is the preferred method for managing diabetes mellitus in Wolfram syndrome. Hypoglycemia alarms should ideally be both audible and vibrating, with adaptation for vision loss if required (consensus ≥80%; level C; Round 2).
Insulin is the mainstay and cornerstone of treatment for diabetes mellitus in Wolfram syndrome. Management of diabetes mellitus should prioritize continuous glucose monitoring integrated with hybrid closed-loop insulin delivery where available64,65. Hypoglycemia alarm systems should incorporate both audible and vibrating alerts, with adaptations for visual impairment.
Statement 21
Children with Wolfram syndrome–related diabetes mellitus should have glycemic control monitored every 3 months (adults every 6 months), using HbA1c, insulin dosing, and continuous glucose monitoring (if available). Pre-diabetic patients should have HbA1c assessed every 4 months. Patients should be taught to seek medical assistance promptly if glucose levels are not well controlled (consensus ≥80%; level B / expert opinion; Round 3).
Glycemic control in Wolfram syndrome-related diabetes mellitus requires regular structured review, incorporating HbA1c, insulin dosing review, and continuous glucose monitoring data where available.
The recommended intervals of every 3 months in children and every 6 months in adults represent minimum review frequencies for clinically stable patients who are meeting their glycemic targets, consistent with the American Diabetes Association Standards of Care66. These intervals are intended as a floor rather than a ceiling. Review should be intensified to at least every 3 months for adults who are not meeting glycemic goals, who have had recent treatment changes, or who experience frequent or severe hypoglycemia or hyperglycemia or other changes in health status, in line with the same guidance.
The 4-month interval recommended for genetically diagnosed individuals who have not yet developed diabetes reflects a different objective than the intervals used for established diabetes. Rather than glycemic management, its purpose is surveillance for conversion, allowing early detection of diabetes onset in patients carrying a confirmed genetic diagnosis of Wolfram syndrome. This approach parallels the established framework for monitoring presymptomatic individuals at risk of diabetes, in which periodic assessment of glycemic status by fasting glucose, oral glucose tolerance testing, and HbA1c is performed at intervals that shorten with higher risk of progression67,68. Because Wolfram syndrome diabetes is non-autoimmune, surveillance relies on these glucose-based measures rather than islet autoantibodies2.
The two sets of intervals therefore serve distinct purposes, surveillance for conversion versus management of established disease, and do not represent a hierarchy of monitoring intensity. Patients and families should be educated to recognize early signs of dysglycemia and to seek medical assistance promptly when glucose levels are not well controlled2.
Statement 22
Arginine vasopressin deficiency (formerly diabetes insipidus) should be diagnosed using paired early morning urine and fasting plasma osmolality and sodium measurements after nocturnal and morning euglycemia. A water deprivation test is a second-line test and should only be undertaken in a specialist center with appropriate supervision (consensus ≥80%; level B / expert opinion; Round 1).
Arginine vasopressin deficiency (formerly known as central diabetes insipidus) results from progressive loss of hypothalamic vasopressin-producing neurons and affects the majority of patients5. It contributes substantially to morbidity through polyuria, polydipsia, and risk of hypernatremic dehydration.
Arginine vasopressin deficiency (formerly central diabetes insipidus) should be diagnosed by measuring paired early-morning urine and plasma osmolality in the setting of euglycemia. Additional neuroendocrine complications, including growth failure, adrenocortical insufficiency, pubertal delay, hypogonadism, and hypothyroidism, should be screened for with targeted investigations as clinically indicated69.
Statement 23
First-line treatment for arginine vasopressin deficiency in Wolfram syndrome is usually oral, sublingual, or intranasal desmopressin (consensus ≥80%; level B / expert opinion; Round 1).
Desmopressin, administered orally, sublingually, or intranasally, is the first-line treatment for arginine vasopressin deficiency. Dosing should be individualized and monitored to avoid hyponatremia, with particular attention to fluid balance during intercurrent illness.
Statement 24
If there are clinical signs of growth failure, adrenocortical insufficiency, pubertal delay, or underactive thyroid, referral to an endocrinologist for anterior pituitary function tests should be considered (consensus ≥80%; expert opinion; Round 1).
Anterior pituitary dysfunction may accompany posterior pituitary involvement. Where there are clinical signs of growth failure, adrenal insufficiency, pubertal delay, or hypothyroidism, referral to an endocrinologist for anterior pituitary function testing should be considered so that hormone deficiencies can be identified and treated70.
Statement 25
To screen for hypogonadism in Wolfram syndrome, take an age-directed history, assess Tanner stage, and measure serum testosterone or estradiol. Second-line tests include FSH, LH, inhibin B, anti-Müllerian hormone, sperm count (males), and dynamic function tests. Management of hypogonadism includes hormone replacement therapy directed by an endocrinologist (consensus ≥80%; expert opinion; Round 1).
Hypogonadism may occur and warrants assessment through an age-directed history, Tanner staging, and measurement of serum testosterone or estradiol. Second-line investigations include FSH, LH, inhibin B, anti-Mullerian hormone, sperm count in males, and dynamic function tests. Hormone replacement therapy, where indicated, should be directed by an endocrinologist71.
Statement 26
Fertility counseling and preservation should be offered in a timely manner if subfertility or infertility is suspected. There are case reports of successful pregnancies and of affected males who have fathered children (consensus ≥80%; level C / expert opinion; Round 1).
Subfertility and infertility may affect some individuals, although successful pregnancies and affected males who have fathered children have been reported. Fertility counseling and preservation should be offered in a timely manner where subfertility or infertility is suspected, ideally before fertility is further compromised72,73.
Statement 27
Height and weight should be monitored in all patients by plotting on standard growth charts. Height should be measured in adults to calculate BMI (consensus ≥80%; expert opinion; Round 2).
Growth and nutritional status should be monitored in all patients by plotting height and weight on standard growth charts, with height measured in adults to calculate body mass index. Deviation from expected growth trajectories should prompt assessment for endocrine and nutritional contributors74-77.
Statement 28
Patients should be screened for dyslipidemia annually if diabetes mellitus is present (consensus ≥80%; expert opinion; Round 1).
Patients with diabetes mellitus are at increased cardiometabolic risk, and dyslipidemia is an important modifiable contributor. Annual lipid screening is recommended where diabetes mellitus is present, with management according to established diabetes care standards78.
Urology and gastroenterology
Statement 29
Baseline urological assessment at diagnosis should include a standardized questionnaire regarding urinary symptoms and a voiding diary; clinical examination and assessment of renal function; blood electrolytes, urea, creatinine, and GFR; bladder and renal ultrasound; and non-invasive urodynamic testing (consensus ≥80%; expert opinion; Round 2).
Neurogenic bladder resulting from autonomic neuropathy affects the majority of patients with advancing disease and carries significant risks of upper urinary tract damage and renal impairment if not identified and managed early.
Neurogenic bladder resulting from autonomic neuropathy is a major source of morbidity in Wolfram syndrome79,80. Urological assessment should commence at diagnosis and include a urinary symptoms questionnaire, voiding diary, clinical examination, serum electrolytes, urea, creatinine, and glomerular filtration rate, a urine albumin-to-creatinine ratio, blood pressure measurement, bladder and renal ultrasound, and non-invasive urodynamic testing. Follow-up urological assessment should occur every 1–2 years or earlier if symptomatic. Clinicians should be aware of acute bladder outflow tract obstruction causing acute kidney injury; there is also an increased risk of urinary tract infections due to urinary stasis.
Statement 30
Within the first year of diagnosis, baseline screening for neurogenic bladder should occur with non-invasive urodynamic function testing, validated questionnaires, and renal ultrasound where indicated. Follow-up screening should occur every 1–2 years (or sooner if symptomatic), at the discretion of the urologist or medical care provider (consensus ≥80%; expert opinion; Round 3).
Baseline screening for neurogenic bladder should be undertaken within the first year of diagnosis using non-invasive urodynamic function testing, validated questionnaires, and renal ultrasound where indicated, complementing the broader baseline urological assessment outlined above by specifying the timing and the recommended follow-up schedule. Follow-up screening every 1-2 years, or sooner if symptomatic, allows early detection of bladder dysfunction before irreversible upper urinary tract damage occurs; the aetiology may relate to a polyuric stretch injury or to autonomic nerve dysfunction79.
Statement 31
Treatment options for neurogenic bladder in Wolfram syndrome should be discussed in conjunction with a pediatric or adult urology team (consensus ≥80%; expert opinion; Round 1).
Treatment of neurogenic bladder should be planned in conjunction with a pediatric or adult urology team. Management depends on whether the bladder is overactive or underactive and typically begins with bladder training, including double voiding, and oral therapeutics; desmopressin may reduce urine output and improve continence. With progression to megacystis and refractory incontinence, clean intermittent self-catheterization is often required, with later options including an indwelling catheter or a Mitrofanoff appendicovesicostomy79.
Statement 32
First-line management of fecal incontinence should include evaluating for and managing constipation-related encopresis or overflow soiling through diet optimization (adequate fiber and water intake), recommending a bowel regimen, and considering pelvic floor physical therapy where possible (consensus ≥80%; expert opinion; Round 3).
Gastrointestinal manifestations including dysmotility, esophageal spasms, and constipation are recognized features of Wolfram syndrome attributable to autonomic neuropathy. Fecal incontinence and bowel dysfunction are increasingly recognized manifestations, arising from autonomic neuropathy affecting the enteric nervous system. Bowel dysfunction in Wolfram syndrome spans a spectrum from constipation and intestinal dysmotility to intractable fecal incontinence, and has a severe impact on quality of life81. Fecal incontinence warrants active clinical inquiry at each review.
Bowel dysfunction, including constipation, intestinal dysmotility, and fecal incontinence, is an underrecognized but clinically important manifestation of Wolfram syndrome attributable to autonomic neuropathy81,82. The spectrum ranges from constipation and intestinal pseudo-obstruction to intractable fecal incontinence, and bowel dysfunction has a severe impact on quality of life. Fecal incontinence, when present, should be evaluated for constipation-related overflow soiling before assuming a primary neurogenic cause. First-line management includes dietary optimization (adequate fiber and fluid intake), a structured bowel regimen, and pelvic floor physical therapy where accessible. Where neurogenic fecal incontinence is confirmed or suspected, specialist gastroenterological assessment should be considered.
Statement 33
Management options for constipation in Wolfram syndrome include diet and behavior advice, stool softeners, fiber supplements, laxatives, and disimpaction regimens (consensus ≥80%; expert opinion; Round 1).
Constipation is common and may reflect autonomic neuropathy affecting the enteric nervous system. Management options include dietary and behavioral advice, stool softeners, fiber supplements, laxatives, and disimpaction regimens, escalated in a stepwise fashion and coordinated with specialist input where symptoms are refractory83.
Psychiatry and cognition
Statement 34
All newly diagnosed patients should be offered assessment by an appropriate healthcare professional (psychologist or psychiatrist) to identify emotional, developmental, and educational needs and to signpost to support (consensus ≥80%; expert opinion; Round 3).
Psychiatric illness, particularly depression, anxiety, and hallucinations, is underrecognized in Wolfram syndrome and contributes substantially to patient burden. Neuropsychiatric conditions including attention-deficit/hyperactivity disorder (ADHD) and obsessive-compulsive disorder (OCD)-related symptoms are recognized and should be assessed as part of routine psychiatric review. Proactive psychiatric assessment and treatment are critical components of care.
Psychiatric illness is one of the most significant and underappreciated sources of morbidity in Wolfram syndrome84,85. Depression, anxiety, and psychosis affect a substantial proportion of patients, and psychiatric symptoms may precede or accompany neurological deterioration. All newly diagnosed patients should be offered assessment by a psychologist or psychiatrist to identify emotional, developmental, and educational needs, and to facilitate access to appropriate support services.
Statement 35
First-line treatment for anxiety and depression in Wolfram syndrome should be psychotherapy and SSRIs, managed in consultation with a psychiatrist. There is limited evidence that fluoxetine and citalopram may be more effective than sertraline; however, other SSRIs can be used as clinically indicated (consensus ≥80%; level C / expert opinion; Round 3).
First-line pharmacological treatment for anxiety and depression is selective serotonin reuptake inhibitor (SSRI) therapy, managed in consultation with a psychiatrist. Limited case series data suggest that fluoxetine and citalopram may be particularly effective in Wolfram syndrome, though other SSRIs remain appropriate when clinically indicated85. Psychotherapy should be offered alongside pharmacological treatment where available.
Discussion
These guidelines represent the first international, multidisciplinary, patient-inclusive clinical consensus for Wolfram syndrome and establish a common framework for diagnosis, monitoring, and management across all major organ systems. They are intended to improve the consistency and quality of clinical care worldwide, including in settings far from specialist centers, where most patients receive their care.
The methodological approach, a three-round international Delphi process informed by a systematic evidence review and incorporating formal patient and family feedback, reflects established best practice in rare disease guideline development32. The achievement of ≥80% consensus on all 35 final statements across 65 international specialists spanning five continents and six clinical disciplines provides a strong foundation of expert agreement. The parallel patient consultation, formally integrated through international advocacy organizations, represents an advance over prior rare disease guideline processes and ensures that the perspectives of those living with Wolfram syndrome are embedded in the recommendations.
Several clinical messages emerge from these guidelines. First, early and accurate genetic diagnosis is essential: Wolfram syndrome is systematically underdiagnosed, and the absence of islet autoantibodies in a child with apparent type 1 diabetes should prompt genetic testing, ideally on a large gene panel so that alternative monogenic causes of diabetes can also be detected, rather than targeted sequencing of WFS1 alone. Second, multidisciplinary coordinated care is mandatory. No single specialty can manage the full clinical complexity of this disease. Third, proactive management of preventable complications, particularly neurogenic bladder, psychiatric illness, and autonomic dysfunction, can substantially reduce morbidity. Fourth, patient advocacy organizations are integral partners in care, not adjuncts.
Palliative and supportive care also merit explicit consideration. Because Wolfram syndrome is progressive and, in its advanced stages, associated with substantial neurological disability and reduced life expectancy, early integration of palliative care can improve quality of life for patients and their families. Conversations about goals of care and advance care planning should be introduced sensitively, revisited as the disease evolves, and embedded within routine multidisciplinary review rather than reserved for times of crisis. Access to symptom management, psychological support, and, where appropriate, hospice services should be considered alongside disease-directed treatment71.
Important limitations should be acknowledged. The evidence base for Wolfram syndrome remains predominantly observational, as there are no randomized controlled trials to guide practice, and most recommendations therefore rest on expert consensus. This reflects both the global rarity of the disease and the challenges of securing research funding for rare conditions. Future iterations of these guidelines should incorporate prospective audit data and, where available, clinical trial evidence, particularly from ongoing therapeutic trials targeting ER stress pathways in Wolfram syndrome.
Future directions
Several disease-modifying strategies are now under active investigation in Wolfram syndrome. Alongside approaches aimed directly at the underlying endoplasmic reticulum stress and mitochondrial dysfunction, repurposing of established diabetes therapies has attracted particular interest86-88.
Limited evidence from preclinical studies and a small number of case reports suggests that glucagon-like peptide-1 (GLP-1) receptor agonists may be beneficial in Wolfram syndrome. Reported effects include reduced insulin requirements and improved glycemic control, with preclinical data raising the possibility that these agents could slow disease progression. A recent case series from Aotearoa, New Zealand describes the phenotype and treatment response in affected individuals89-94. In preclinical models, GLP-1 receptor agonist therapy has been reported to ameliorate visual neurodegeneration and sensorineural hearing loss, raising the possibility of benefits beyond glycemic control, although this has not been demonstrated in patients92,95. These agents are currently one possible adjunctive option rather than a replacement for insulin, and should not completely replace insulin in children. Their known gastrointestinal side effects must be carefully considered in the context of existing autonomic dysfunction and growth delay in some patients.
These guidelines provide a comprehensive framework for improving the diagnosis and clinical management of Wolfram syndrome internationally. Coordinated multidisciplinary care, early genetic diagnosis, and proactive management of evolving complications are the pillars of good clinical practice for this disease. Implementation of these recommendations should be accompanied by a prospective audit to expand the evidence base and support future updates. With a growing pipeline of therapeutic candidates targeting ER stress and mitochondrial dysfunction in Wolfram syndrome, there is an increasingly urgent need to establish international networks that can rapidly develop and validate the outcomes that matter most to patients, deliver consistent best supportive care today, and provide high-quality infrastructure for future randomized controlled trials.
Acknowledgements
The authors express their sincere thanks to the participating patient support and advocacy organizations and to the patients and families whose input shaped these recommendations.
Funding
FU is partly supported by grants from the National Institutes of Health (NIH)/NIDDK (DK132090 and DK020579). Research reported in this publication was also partly supported by the Washington University Institute of Clinical and Translational Sciences through grant UL1TR002345 from the NIH/NCATS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
PYWM is supported by an Advanced Fellowship Award (NIHR301696) from the UK National Institute for Health and Care Research (NIHR). PYWM also receives funding from the Rosetrees Trust (PGL23/100048), Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity (GR001376), the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the International Foundation for Optic Nerve Disease (IFOND), the NIHR as part of the Rare Diseases Translational Research Collaboration, the NIHR Cambridge Biomedical Research Centre (NIHR203312), and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (NIHR203322). This research was supported by LifeArc under grant no. 10748; LifeArc is a charity registered in England and Wales under no. 1015243 and in Scotland under no. SC037861. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
MS was supported by the National Institute for Health and Care Research Exeter Biomedical Research Centre and the National Institute for Health and Care Research Exeter Clinical Research Facility. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
AR’s effort on this project was supported in part by the Foundation for Barnes-Jewish Hospital (grant number 6898).
Author Contributions
FU and TB conceived and co-chaired the study and designed the modified three-round Delphi methodology. FU, TB, JE, SA, PYWM, SG, SGe, and TL constituted the international steering committee and together conducted the systematic literature review, drafted the candidate consensus statements across the six clinical domains, and iteratively revised the statements between voting rounds. SG, SGe, and TL led patient and family engagement and coordinated the parallel patient and caregiver survey through the participating international advocacy organizations. The members of the International Wolfram Syndrome Clinical Guidelines Consortium contributed to the modified three-round Delphi process in several ways, including rating candidate statements, providing structured feedback and suggestions to refine the statements, and contributing to the writing, review, and editing of the manuscript. JE, SA, FU, and TB analyzed the Delphi data and wrote the original draft. All authors and all members of the International Wolfram Syndrome Clinical Guidelines Consortium critically reviewed the manuscript and approved the final version.
Competing Interests
FU has a sponsored research agreement with and received material support from Prilenia Therapeutics; has received National Institutes of Health (NIH) grants, royalties from Novus Biologicals and Sana Biotechnology, licensing and/or consulting fees from Opris Biotechnologies and Emerald Biotherapeutics, and travel fees from Wolfram France, Wolfram UK, and the Snow Foundation; serves in unpaid advisory roles for the Snow Foundation and Be A Tiger Foundation; holds US patents 9,891,231, 10,441,574, and 10,695,324; and was President and shareholder of the now-dissolved CURE4WOLFRAM. BM reports salary support from Amylyx Pharmaceuticals and serves as an unpaid member of the American Academy of Pediatrics Section on Endocrinology Executive Committee. SA reports salary support from an NIH KL2 Award, research support from Uplifting Athletes, and travel support from the Epilepsy Foundation. TH reports salary support from Amylyx, NIH grants, honoraria and travel fees from the University of Southern California, and honoraria for grant reviews from the Clayco Foundation. IBH reports a research grant from COUR Pharmaceuticals and consulting relationships with Abbott, Roche, and Hagar. PYWM reports consultancy work for GenSight Biologics, Chiesi, PYC Therapeutics, and Stoke Therapeutics. SIS serves as an advisory board member for Zealand Pharmaceuticals and MannKind Inc. and as a research investigator for Rhythm Pharmaceuticals Inc.
Figures
Figure 1. Modified Delphi process for the development of international consensus clinical guidelines for Wolfram syndrome.
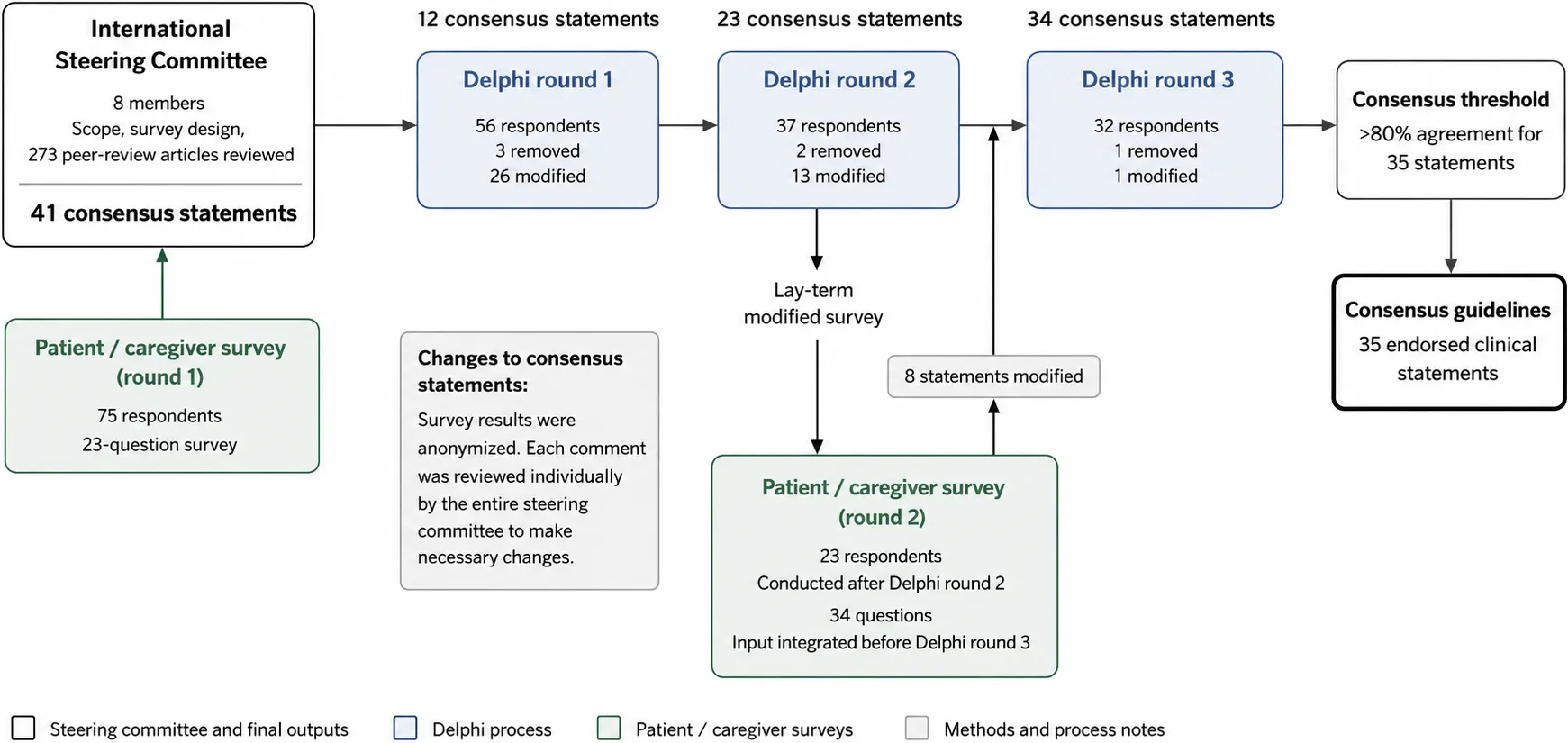
An international steering committee of 8 members defined the scope and survey design based on a review of 273 peer-reviewed manuscripts and input from patients and caregivers (23-question survey). A three-round Delphi survey using a 5-point Likert scale, and an optional space for comments for each statement, was administered to an international expert panel (Round 1: n = 56; Round 2: n = 37; Round 3: n = 32 respondents). Between rounds, statements were modified or removed based on comments or if they did not reach the pre-defined consensus threshold of 80% (Round 1: 26 modified, 3 removed; Round 2: 13 modified, 2 removed; Round 3: 1 modified, 1 removed). For each revision, the steering committee discussed every free-text comment from survey respondents. A parallel patient/caregiver survey (34 lay-term questions, conducted after Round 2; 23 respondents) was integrated before Round 3. Survey results were anonymized, and each comment from patient/caregiver was also reviewed individually by the steering committee. The entire process yielded 35 endorsed clinical consensus statements with > 80% agreement.
Figure 2. Diagnostic algorithm for Wolfram Spectrum Disorders.
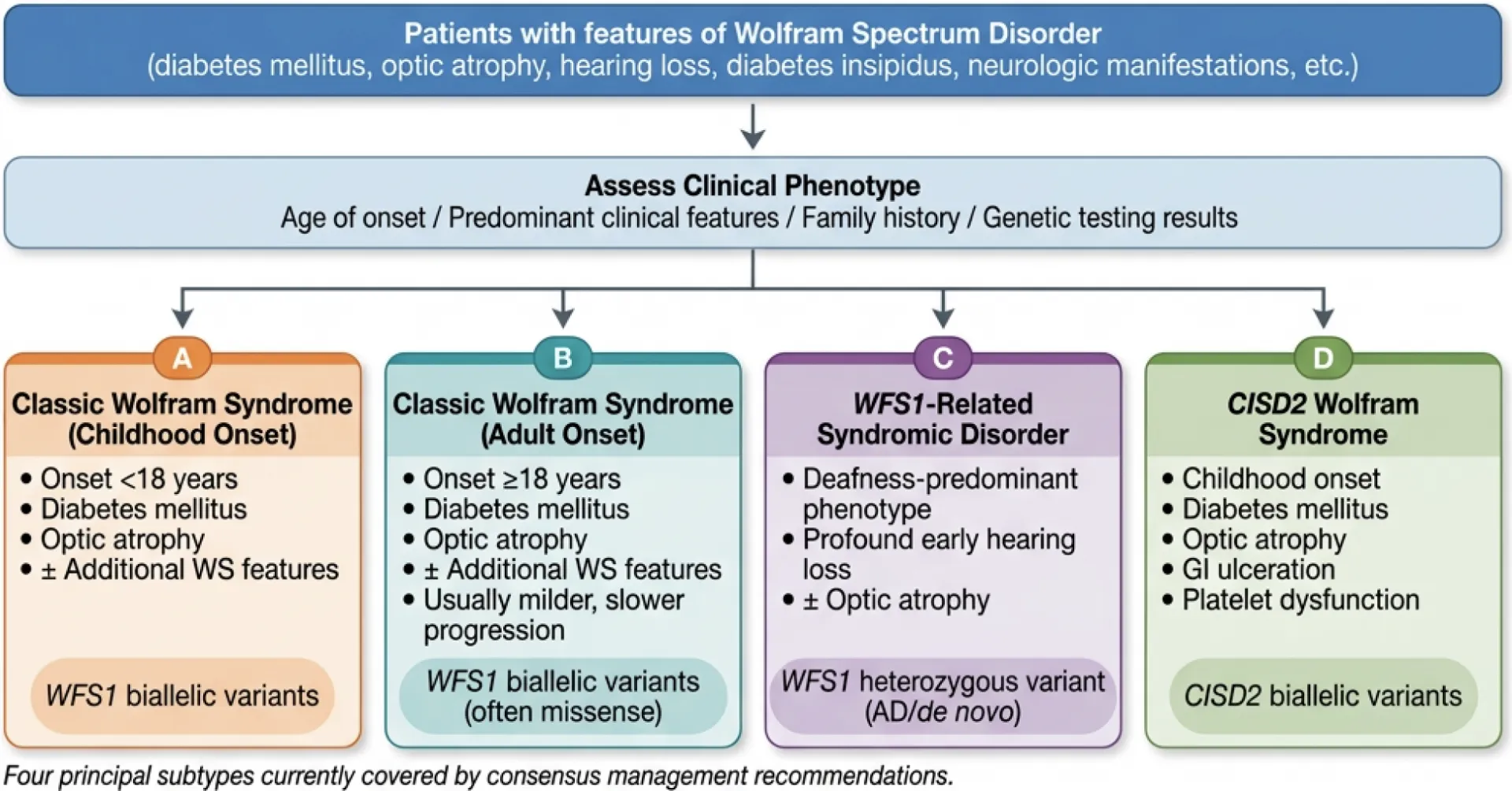
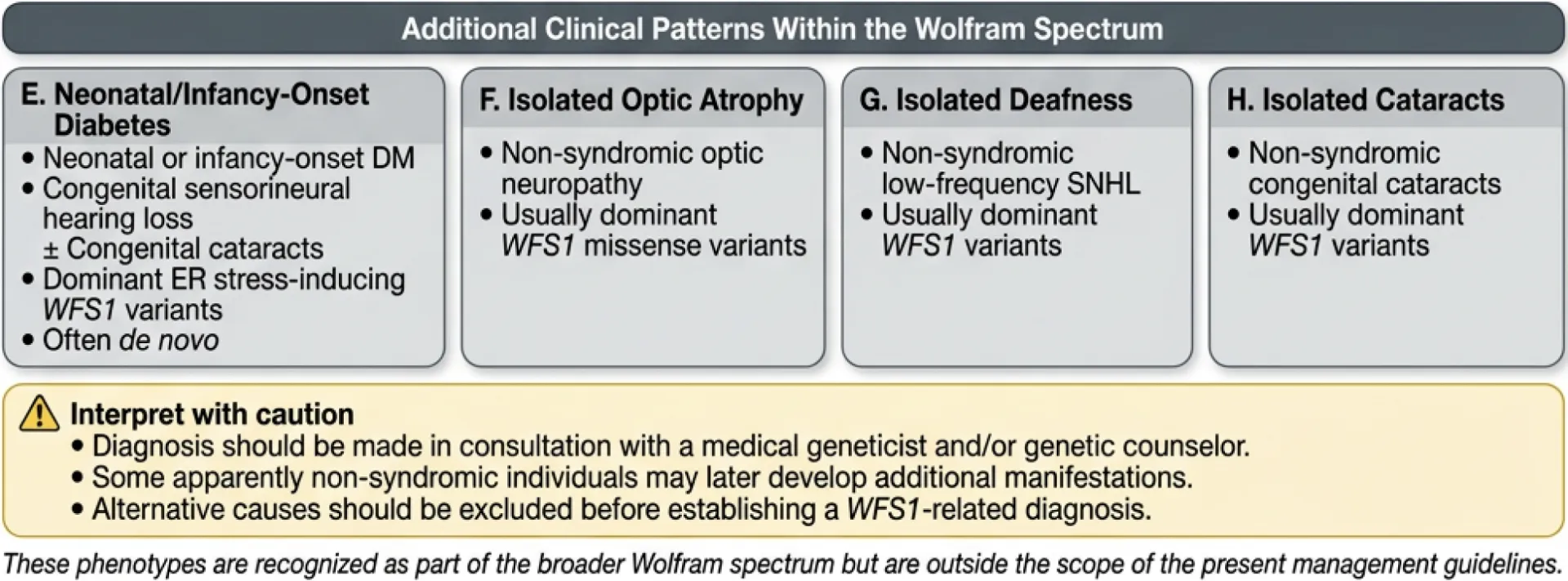
Patients are classified into one of four principal Wolfram syndrome subtypes (A–D) based on age of onset, predominant clinical features, and genetic findings. Four additional WFS1-related clinical patterns (E–H), including neonatal/infancy-onset disease and isolated optic atrophy, are shown to represent the full eight-pattern Wolfram spectrum disorder; patterns E–H fall outside the scope of these management guidelines.
Table 1. Patient advocacy organizations involved in the development of these guidelines.
Affiliations
- Department of Cancer and Genomic Sciences, University of Birmingham, United Kingdom
- Department of Neurology, Washington University School of Medicine, St. Louis, Missouri, USA
- John van Geest Centre for Brain Repair and MRC Mitochondrial Biology Unit, Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- Cambridge Eye Unit, Addenbrooke’s Hospital, Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
- Moorfields Eye Hospital NHS Foundation Trust, London, United Kingdom
- Institute of Ophthalmology, University College London, London, United Kingdom
- The Snow Foundation, St. Louis, USA
- Wolfram Syndrome UK, United Kingdom
- Department of Medicine, Division of Endocrinology, Metabolism, and Lipid Research, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110, USA
- Department of Pathology and Immunology, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110, USA
References
- Wolfram DJ, Wagener HP. Diabetes mellitus and simple optic atrophy among siblings: report of four cases. May Clin Proc 1938; 1: 715-8.
- Urano F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Current diabetes reports 2016; 16(1): 6.
- Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nature genetics 1998; 20(2): 143-8.
- Amr S, Heisey C, Zhang M, et al. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. American journal of human genetics 2007; 81(4): 673-83.
- Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346(8988): 1458-63.
- Lee EM, Verma M, Palaniappan N, et al. Genotype and clinical characteristics of patients with Wolfram syndrome and WFS1-related disorders. Front Genet 2023; 14: 1198171.
- Lesperance MM, Hall JW, 3rd, San Agustin TB, Leal SM. Mutations in the Wolfram syndrome type 1 gene (WFS1) define a clinical entity of dominant low-frequency sensorineural hearing loss. Archives of otolaryngology–head & neck surgery 2003; 129(4): 411-20.
- Roberts JP, Tang AF, Hernandez D, et al. Natural history of 15 patients with autosomal dominant WFS1 pathogenic variants associated with sensorineural hearing loss and optic atrophy. Orphanet journal of rare diseases 2026.
- De Franco E, Flanagan SE, Yagi T, et al. Dominant ER Stress-Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes 2017; 66(7): 2044-53.
- Kline AD, Moss JF, Selicorni A, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nature reviews Genetics 2018; 19(10): 649-66.
- Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet neurology 2018; 17(3): 251-67.
- Korbonits M, Blair JC, Boguslawska A, et al. Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: Part 1, general recommendations. Nature reviews Endocrinology 2024; 20(5): 278-89.
- Brioude F, Kalish JM, Mussa A, et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nature reviews Endocrinology 2018; 14(4): 229-49.
- Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018; 28(2): 103-15.
- Tahani N, Maffei P, Dollfus H, et al. Consensus clinical management guidelines for Alstrom syndrome. Orphanet journal of rare diseases 2020; 15(1): 253.
- Diamond IR, Grant RC, Feldman BM, et al. Defining consensus: a systematic review recommends methodologic criteria for reporting of Delphi studies. J Clin Epidemiol 2014; 67(4): 401-9.
- Strom TM, Hortnagel K, Hofmann S, et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998; 7(13): 2021-8.
- Hofmann S, Philbrook C, Gerbitz KD, Bauer MF. Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 2003; 12(16): 2003-12.
- Takeda K, Inoue H, Tanizawa Y, et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001; 10(5): 477-84.
- Fonseca SG, Fukuma M, Lipson KL, et al. WFS1 Is a Novel Component of the Unfolded Protein Response and Maintains Homeostasis of the Endoplasmic Reticulum in Pancreatic Beta-Cells. The Journal of biological chemistry 2005; 280(47): 39609-15.
- Astuti D, Sabir A, Fulton P, et al. Monogenic diabetes syndromes: Locus-specific databases for Alstrom, Wolfram, and Thiamine-responsive megaloblastic anemia. Human mutation 2017; 38(7): 764-77.
- Cryns K, Sivakumaran TA, Van den Ouweland JM, et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Human mutation 2003; 22(4): 275-87.
- Khanim F, Kirk J, Latif F, Barrett TG. WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Human mutation 2001; 17(5): 357-67.
- de Heredia ML, Cleries R, Nunes V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 2013; 15(7): 497-506.
- Fonseca SG, Ishigaki S, Oslowski CM, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. The Journal of clinical investigation 2010; 120(3): 744-55.
- Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. The EMBO journal 2010; 29(3): 606-18.
- Chen YF, Kao CH, Chen YT, et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes & development 2009; 23(10): 1183-94.
- Tamir S, Paddock ML, Darash-Yahana-Baram M, et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochimica et biophysica acta 2015; 1853(6): 1294-315.
- Ateya R, Ciecierega T, Abusamra M, Abuawwad M, Abu-Libdeh A, Sultan M. Wolfram Syndrome-2, a Cause of Severe Gastrointestinal Bleeding: A Case Series and a Literature Review. JPGN Rep 2023; 4(3): e339.
- Grenier J, Meunier I, Daien V, et al. WFS1 in Optic Neuropathies: Mutation Findings in Nonsyndromic Optic Atrophy and Assessment of Clinical Severity. Ophthalmology 2016; 123(9): 1989-98.
- Krutish A, Elmore J, Ilse W, et al. A novel WFS1 variant associated with isolated congenital cataracts. Cold Spring Harb Mol Case Stud 2023; 9(1).
- Eiberg H, Hansen L, Kjer B, et al. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet 2006; 43(5): 435-40.
- Rendtorff ND, Lodahl M, Boulahbel H, et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. American journal of medical genetics Part A 2011; 155A(6): 1298-313.
- Barrett TG, Bundey SE. Wolfram (DIDMOAD) syndrome. J Med Genet 1997; 34(10): 838-41.
- Bespalova IN, Van Camp G, Bom SJ, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 2001; 10(22): 2501-8.
- Kitamura RA, Maxwell KG, Ye W, et al. Multidimensional analysis and therapeutic development using patient iPSC-derived disease models of Wolfram syndrome. JCI Insight 2022; 7(18).
- Wilf-Yarkoni A, Shor O, Fellner A, et al. Mild Phenotype of Wolfram Syndrome Associated With a Common Pathogenic Variant Is Predicted by a Structural Model of Wolframin. Neurol Genet 2021; 7(2): e578.
- Bansal V, Boehm BO, Darvasi A. Identification of a missense variant in the WFS1 gene that causes a mild form of Wolfram syndrome and is associated with risk for type 2 diabetes in Ashkenazi Jewish individuals. Diabetologia 2018; 61(10): 2180-8.
- Bonnycastle LL, Chines PS, Hara T, et al. Autosomal dominant diabetes arising from a wolfram syndrome 1 mutation. Diabetes 2013; 62(11): 3943-50.
- Sriram A, Wakeling MN, Hattersley AT, et al. Rare Variants in NEUROD1 and PDX1 Are Low-Penetrance Causes of MODY, Whereas Those in APPL1 and WFS1 Are Not Associated With MODY. Diabetes 2025; 74(11): 2123-31.
- Grzybowska-Adamowicz J, Gadzalska K, Jakiel P, et al. Patients with a Wide Range of Disorders Related to WFS1 Gene Variants: Novel Mutations and Genotype-Phenotype Correlations. Genes (Basel) 2024; 15(12).
- Cleverley K, Davies J, Allemang B, Brennenstuhl S. Validation of the Transition Readiness Assessment Questionnaire (TRAQ) 5.0 for use among youth in mental health services. Child Care Health Dev 2023; 49(2): 248-57.
- Nagra A, McGinnity PM, Davis N, Salmon AP. Implementing transition: Ready Steady Go. Arch Dis Child Educ Pract Ed 2015; 100(6): 313-20.
- Hoekel J, Chisholm SA, Al-Lozi A, Hershey T, Tychsen L, Washington University Wolfram Study G. Ophthalmologic correlates of disease severity in children and adolescents with Wolfram syndrome. Journal of AAPOS 2014; 18(5): 461-5 e1.
- Hoekel J, Narayanan A, Rutlin J, et al. Visual pathway function and structure in Wolfram syndrome: patient age, variation and progression. BMJ Open Ophthalmol 2018; 3(1): e000081.
- McNeely BJ, Tang AF, Urano F, et al. Optic Atrophy in Wolfram Syndrome Type 1: A Retrospective Analysis of Visual Outcomes and Biomarker Correlates. Journal of neuro-ophthalmology 2026.
- Hershey T, Lugar HM, Shimony JS, et al. Early Brain Vulnerability in Wolfram Syndrome. PloS one 2012; 7(7): e40604.
- Samara A, Rahn R, Neyman O, et al. Developmental hypomyelination in Wolfram syndrome: new insights from neuroimaging and gene expression analyses. Orphanet journal of rare diseases 2019; 14(1): 279.
- Samara A, Lugar HM, Hershey T, Shimony JS. Longitudinal Assessment of Neuroradiologic Features in Wolfram Syndrome. AJNR Am J Neuroradiol 2020; 41(12): 2364-9.
- Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol 1989; 46(10): 1121-3.
- Plantinga RF, Pennings RJ, Huygen PL, et al. Hearing impairment in genotyped Wolfram syndrome patients. Ann Otol Rhinol Laryngol 2008; 117(7): 494-500.
- Karzon RK, Hullar TE. Audiologic and vestibular findings in Wolfram syndrome. Ear and hearing 2013; 34(6): 809-12.
- Karzon RK, Hullar TE. Audiologic and vestibular findings in Wolfram syndrome. Ear and hearing 2013; 34(6): 809-12.
- Kinsley BT, Swift M, Dumont RH, Swift RG. Morbidity and mortality in the Wolfram syndrome. Diabetes care 1995; 18(12): 1566-70.
- Rommel N, Hamdy S. Oropharyngeal dysphagia: manifestations and diagnosis. Nat Rev Gastroenterol Hepatol 2016; 13(1): 49-59.
- Licis A, Davis G, Eisenstein SA, Lugar HM, Hershey T. Sleep disturbances in Wolfram syndrome. Orphanet journal of rare diseases 2019; 14(1): 188.
- Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res 2011; 21(2): 69-72.
- Marshall BA, Permutt MA, Paciorkowski AR, et al. Phenotypic characteristics of early Wolfram syndrome. Orphanet journal of rare diseases 2013; 8(1): 64.
- Doty T, Foster ER, Marshall B, Ranck S, Hershey T. The effects of disease-related symptoms on daily function in Wolfram Syndrome. Transl Sci Rare Dis 2017; 2(1-2): 89-100.
- Waschbisch A, Volbers B, Struffert T, Hoyer J, Schwab S, Bardutzky J. Primary diagnosis of Wolfram syndrome in an adult patient–case report and description of a novel pathogenic mutation. Journal of the neurological sciences 2011; 300(1-2): 191-3.
- Chaussenot A, Bannwarth S, Rouzier C, et al. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Annals of neurology 2011; 69(3): 501-8.
- Ray MK, Chen L, White NH, Ni R, Hershey T, Marshall BA. Longitudinal progression of diabetes mellitus in Wolfram syndrome: The Washington University Wolfram Research Clinic experience. Pediatric diabetes 2022; 23(2): 212-8.
- Libman I, Haynes A, Lyons S, et al. ISPAD Clinical Practice Consensus Guidelines 2022: Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatric diabetes 2022; 23(8): 1160-74.
- Rohayem J, Ehlers C, Wiedemann B, et al. Diabetes and neurodegeneration in Wolfram syndrome: a multicenter study of phenotype and genotype. Diabetes care 2011; 34(7): 1503-10.
- Zmyslowska A, Fendler W, Szadkowska A, et al. Glycemic variability in patients with Wolfram syndrome is lower than in type 1 diabetes. Acta diabetologica 2015; 52(6): 1057-62.
- American Diabetes Association Professional Practice Committee for D. 6. Glycemic Goals, Hypoglycemia, and Hyperglycemic Crises: Standards of Care in Diabetes-2026. Diabetes care 2026; 49(Supplement_1): S132-S49.
- Phillip M, Achenbach P, Addala A, et al. Consensus Guidance for Monitoring Individuals With Islet Autoantibody-Positive Pre-Stage 3 Type 1 Diabetes. Diabetes care 2024; 47(8): 1276-98.
- Besser REJ, Bell KJ, Couper JJ, et al. ISPAD Clinical Practice Consensus Guidelines 2022: Stages of type 1 diabetes in children and adolescents. Pediatric diabetes 2022; 23(8): 1175-87.
- Fleseriu M, Hashim IA, Karavitaki N, et al. Hormonal Replacement in Hypopituitarism in Adults: An Endocrine Society Clinical Practice Guideline. The Journal of clinical endocrinology and metabolism 2016; 101(11): 3888-921.
- Medlej R, Wasson J, Baz P, et al. Diabetes mellitus and optic atrophy: a study of Wolfram syndrome in the Lebanese population. The Journal of clinical endocrinology and metabolism 2004; 89(4): 1656-61.
- Frontino G, Delvecchio M, Prudente S, et al. SID/SIEDP expert consensus on optimizing clinical strategies for early detection and management of wolfram syndrome. J Endocrinol Invest 2025; 48(3): 507-25.
- Caretto A, Scarascia Mugnozza F, Valsecchi F, et al. Diabetes mellitus and pregnancy in Wolfram syndrome type 1: a case report with review of clinical and pathophysiological aspects. Front Med (Lausanne) 2025; 12: 1639884.
- Haghighi A, Haghighi A, Setoodeh A, Saleh-Gohari N, Astuti D, Barrett TG. Identification of homozygous WFS1 mutations (p.Asp211Asn, p.Gln486*) causing severe Wolfram syndrome and first report of male fertility. European journal of human genetics 2013; 21(3): 347-51.
- Sundberg F, deBeaufort C, Krogvold L, et al. ISPAD Clinical Practice Consensus Guidelines 2022: Managing diabetes in preschoolers. Pediatric diabetes 2022; 23(8): 1496-511.
- Serbis A, Rallis D, Giapros V, Galli-Tsinopoulou A, Siomou E. Wolfram Syndrome 1: A Pediatrician’s and Pediatric Endocrinologist’s Perspective. International journal of molecular sciences 2023; 24(4).
- de Onis M, Onyango AW, Borghi E, Siyam A, Nishida C, Siekmann J. Development of a WHO growth reference for school-aged children and adolescents. Bull World Health Organ 2007; 85(9): 660-7.
- Kuczmarski RJ, Ogden CL, Guo SS, et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11 2002; (246): 1-190.
- American Diabetes Association Professional Practice Committee for D. 10. Cardiovascular Disease and Risk Management: Standards of Care in Diabetes-2026. Diabetes care 2026; 49(Supplement_1): S216-S45.
- Wragg R, Dias RP, Barrett T, McCarthy L. Bladder dysfunction in Wolfram syndrome is highly prevalent and progresses to megacystis. J Pediatr Surg 2018; 53(2): 321-5.
- La Valle A, Piccolo G, Maghnie M, d’Annunzio G. Urinary Tract Involvement in Wolfram Syndrome: A Narrative Review. Int J Environ Res Public Health 2021; 18(22).
- Orssaud C. Foecal incontinence disorders in Wolfram syndrome: a new manifestation. J Med Genet 2025; 62(2): 107-8.
- Liu Z, Sakakibara R, Uchiyama T, et al. Bowel dysfunction in Wolfram syndrome. Diabetes care 2006; 29(2): 472-3.
- Chang L, Chey WD, Imdad A, et al. American Gastroenterological Association-American College of Gastroenterology Clinical Practice Guideline: Pharmacological Management of Chronic Idiopathic Constipation. Am J Gastroenterol 2023; 118(6): 936-54.
- Bischoff AN, Reiersen AM, Buttlaire A, et al. Selective cognitive and psychiatric manifestations in Wolfram Syndrome. Orphanet journal of rare diseases 2015; 10: 66.
- Reiersen AM, Noel JS, Doty T, Sinkre RA, Narayanan A, Hershey T. Psychiatric Diagnoses and Medications in Wolfram Syndrome. Scand J Child Adolesc Psychiatr Psychol 2022; 10(1): 163-74.
- Urano F, Marshall BA, Hurst S, et al. Phase II trial of sodium phenylbutyrate and taurursodiol in Wolfram syndrome. The Journal of clinical investigation 2026; 136(10).
- Dias RP, Brock K, Hu K, et al. Sodium valproate, a potential repurposed treatment for the neurodegeneration in Wolfram syndrome (TREATWOLFRAM): trial protocol for a pivotal multicentre, randomised double-blind controlled trial. BMJ Open 2025; 15(2): e091495.
- Abreu D, Stone SI, Pearson TS, et al. A phase Ib/IIa clinical trial of dantrolene sodium in patients with Wolfram syndrome. JCI Insight 2021; 6(15).
- Gorgogietas V, Rajaei B, Heeyoung C, et al. GLP-1R agonists demonstrate potential to treat Wolfram syndrome in human preclinical models. Diabetologia 2023; 66(7): 1306-21.
- Kondo M, Tanabe K, Amo-Shiinoki K, et al. Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 2018; 61(10): 2189-201.
- Frontino G, Raouf T, Canarutto D, et al. Case Report: Off-Label Liraglutide Use in Children With Wolfram Syndrome Type 1: Extensive Characterization of Four Patients. Front Pediatr 2021; 9: 755365.
- Jagomae T, Seppa K, Reimets R, et al. Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype. Cells 2021; 10(11).
- Png D, Yeoh E, Tan C, Lim SC. A Pair of Siblings With Wolfram Syndrome: A Review of the Literature and Treatment Options. J Investig Med High Impact Case Rep 2023; 11: 23247096221150631.
- L’Amie A, Vincent AL, Jefferies CA, et al. Wolfram syndrome and diabetes mellitus in Aotearoa, New Zealand: Phenotype and response to GLP-1 receptor agonist therapy. Diabetic medicine 2026: e70405.
- Seppa K, Toots M, Reimets R, et al. GLP-1 receptor agonist liraglutide has a neuroprotective effect on an aged rat model of Wolfram syndrome. Scientific reports 2019; 9(1): 15742.